 August
11
August
11
Tags
Alzheimer’s disease: back to the basics & exploring new frontiers
“You, your joys and your sorrows, your memories and ambitions, your sense of personal identity and free will, are in fact no more than the behavior of a vast assembly of nerve cells and their associated molecules. ” –Francis Crick
Watching my grandfather slowly fall prey to Alzheimer’s disease, I was struck by how intricately our memories are entwined with our identities. Cliché, I know. The se ntiment that “we are our memories” has been repeated so often that it has lost some of its emotional force. But no matter how many times you have heard it said, nothing can prepare you for the reality of actually watching someone’s memories slowly slip away, of watching an incredibly sharp man—both in intellect and in humor—become lonely and agitated and frightened, no longer able to recognize the people who care for him day after day. There is something uniquely terrifying in imagining not knowing the people that you yourself hold most dearly, not remembering the events, both joyful and tragic, that have shaped the person you’ve become.
ntiment that “we are our memories” has been repeated so often that it has lost some of its emotional force. But no matter how many times you have heard it said, nothing can prepare you for the reality of actually watching someone’s memories slowly slip away, of watching an incredibly sharp man—both in intellect and in humor—become lonely and agitated and frightened, no longer able to recognize the people who care for him day after day. There is something uniquely terrifying in imagining not knowing the people that you yourself hold most dearly, not remembering the events, both joyful and tragic, that have shaped the person you’ve become.
When my grandfather was diagnosed with Alzheimer’s disease (AD), I wanted to better understand what exactly was happening in his brain as his memory slowly deteriorated. Perhaps this drive came from innate scientific curiosity, or perhaps burying myself in the gritty molecular details was my attempt at escaping from the emotional gravity of the situation. Whatever the motivating force, as a neurobiology major in college, I had the background to understand the gist of most AD research publications. I read article after article. After graduating, I worked for two years in Dr. Karen Duff’s laboratory studying the spread of pathology in AD. While I no longer work directly on AD, two big publications this year from the Duff lab inspired me to revisit the literature in an attempt to explain what is known about how AD starts and progresses, as well as to delve into some of the most interesting new research that has come out this year.
 AD pathology is often described as a combination of “plaques and tangles.” Two molecules, beta amyloid (Aβ) and tau, take center stage in most AD research. Both of them exist in the healthy brain, but in AD they start to cluster, wreaking havoc. Aβ accumulates outside neurons while tau accumulates inside neurons. Aβ clusters are called “plaques” and tau clusters are the “tangles.”
AD pathology is often described as a combination of “plaques and tangles.” Two molecules, beta amyloid (Aβ) and tau, take center stage in most AD research. Both of them exist in the healthy brain, but in AD they start to cluster, wreaking havoc. Aβ accumulates outside neurons while tau accumulates inside neurons. Aβ clusters are called “plaques” and tau clusters are the “tangles.”
Why do plaques and tangles affect memory? It’s not the plaques or tangles themselves that are memory-specific, but they start to accumulate in the hippocampus, the memory center of the brain.  As the hippocampal cells become damaged and die, the brain loses its ability to form and recall memories. Scientists still do not know what makes the hippocampus particularly vulnerable to the formation of these clusters. Eventually the clusters spread from the hippocampus throughout the brain, which is why, in later stages of disease, an AD patient often has not only dementia but also an altered personality or mood and trouble speaking and moving. While some scientists think the focus should be Aβ and others think the target should be tau, AD researchers share a common goal: stopping the death of neurons.
As the hippocampal cells become damaged and die, the brain loses its ability to form and recall memories. Scientists still do not know what makes the hippocampus particularly vulnerable to the formation of these clusters. Eventually the clusters spread from the hippocampus throughout the brain, which is why, in later stages of disease, an AD patient often has not only dementia but also an altered personality or mood and trouble speaking and moving. While some scientists think the focus should be Aβ and others think the target should be tau, AD researchers share a common goal: stopping the death of neurons.
A Catastrophic Cascade
The most widely accepted notion of how AD develops is called the “amyloid cascade hypothesis.” The cascade hypothesis (generally supported by decades of research) posits that an unclear combination of genetic risk factors and environmental stressors leads to the accumulation of a specific type of Aβ that more easily forms clusters than other types. This is thought to begin occurring in a patient’s brain about 10-20 years before any symptoms are evident! The brain is equipped with machinery to clear anything—like Aβ clusters—that shouldn’t be there, but over time this machinery tires and becomes less efficient, and clusters get bigger. As the situation escalates, the brain’s alarm system sounds. The alarm sends the “first responder” cells of the brain—called microglia—into an activated state.
While microglia are important for helping to clear plaques, they can be harmful as well. Imagine a bunch of fire trucks summoned to put out a fire in an apartment building. As multiple hoses fight the source of the fire, the strong streams of water end up damaging many of the surrounding apartments. In a way, this is what happens when the brain tries to fight plaques. The activated microglia release substances that end up stressing out the surrounding brain cells. This is an issue you’ve undoubtedly heard of: inflammation. Immune cells rush to a site of injury or disease to help fix the problem but end up causing some nasty collateral damage.
Chronic inflammation and perhaps also the plaques themselves lead to a host of problems for the neurons. At the top of that list is an alteration in the healthy tau inside the cells, causing it—like the Aβ—to form clusters, further stressing the neurons. The synapses—the points at which two neurons communicate with each other—are destroyed. These points of communication are necessary for any information (including memories!) to travel through the brain, so as synapses are destroyed, brain function is altered. Eventually, the neurons themselves die. As mentioned before, this molecular disaster starts in the hippocampus and eventually spreads throughout the brain.
When considering the amyloid cascade hypothesis, it is important to note that many people with high levels of Aβ have no widespread cell death or cognitive deficits [1]. While Aβ is commonly thought to be the initiator of AD pathology, it is not the sole culprit. Without the rest of the cascade, particularly the buildup of tangles and death of neurons, there is no AD.
While there are still many aspects of AD that we do not understand, there is an incredible task force of researchers focused on learning all there is to know and continuously churning out new research. Just in the last few months, for example, a series of papers in high profile journals have shed some more light on microglia, tau, and viruses in AD.
The Double Life of Microglia
This past March, Dr. Beth Steven’s group at Harvard published a surprising study about the role of microglia in AD [2].  Microglia—the first responders mentioned before—have long been known to help clear plaques and unfortunately lead to a chronic inflammatory state as they take on the valiant task of eradicating plaques. This new study, however, suggests that microglia might be more malicious than we thought, devouring synapses at frighteningly early stages of the disease before plaques have formed. It suggests that, before they even start to form insoluble plaques, small soluble clusters of Aβ cause neurons to wave a white flag that attracts the microglia, which then gobble up the synapses. Remember, synapses are the communication points between two neurons that allow for the transmission of information through the brain. The results in this publication lead to the conclusion that microglia and a common signaling system that microglia use to interact with neurons (called the “complement system”) might be useful drug targets.
Microglia—the first responders mentioned before—have long been known to help clear plaques and unfortunately lead to a chronic inflammatory state as they take on the valiant task of eradicating plaques. This new study, however, suggests that microglia might be more malicious than we thought, devouring synapses at frighteningly early stages of the disease before plaques have formed. It suggests that, before they even start to form insoluble plaques, small soluble clusters of Aβ cause neurons to wave a white flag that attracts the microglia, which then gobble up the synapses. Remember, synapses are the communication points between two neurons that allow for the transmission of information through the brain. The results in this publication lead to the conclusion that microglia and a common signaling system that microglia use to interact with neurons (called the “complement system”) might be useful drug targets.
Fire up the Neuronal Pac-Men
While microglia are cleaning up plaques and responding to (or causing!) problems around neurons, neurons have multiple systems in place for making sure that the inside of the cell is clean and functioning properly. In one of these systems, a few molecules run around labeling anything that is old or malfunctioning or no longer needed. Literally, they put a little tag called “ubiquitin” on anything that needs to be degraded. The tag is recognized by a proteasome, which you can imagine as a neuronal Pac-Man that promptly consumes anything with that ubiquitin tag. The AD field had already shown on multiple occasions that clusters of tau slow the entire proteasome system, but this year Dr. Karen Duff’s group published a new way to rev the system. Rejuvenating the Pac-Men helped to clear tau clusters in an AD mouse model, and the mice performed better in a spatial learning test after receiving the treatment [3].
Please Be Less Excited
In the past few years, a handful of research groups have presented evidence that tau clusters can spread from neuron to neuron via synapses. Another study from Dr. Duff’s lab published last month helped to further confirm this theory. The group used a fancy tool called a microfluidic chamber with three compartments. There were neuron cell bodies in each compartment, but the liquid in each compartment was kept totally separate. The only connections between the compartments were the long, thin cellular extensions emerging from the cell bodies, the tips of which form a synapses with the cells in the next compartment. Tau clusters were initiated only in the first compartment but spread from the first compartment to the second and from the second to the third via these synapses [4].
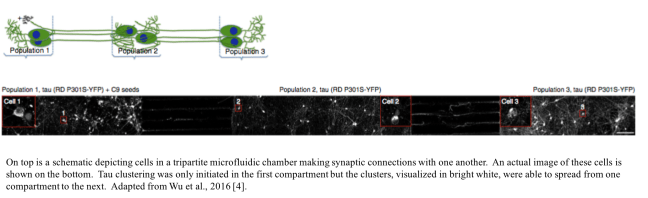
It gets scarier. The study also showed that neuronal activity (neurons talking to each other, essentially the basis of any thought or action you might perform) increased the speed with which the tau clusters spread from cell to cell. This was true both in a mouse model and in cells in a dish [4]. What does this mean? No, being a couch potato with no thoughts or feelings will not help you avoid AD—mental and physical activity help to strengthen synapses against potential disease and dysfunction later in life. However, these results may mean that drugs that decrease neurons’ excitability might be helpful if used before the tau pathology can spread too far.
Can Herpes Cause Alzheimer’s?!
Probably the most surprising AD-related finding of the past few months was a study proposing and describing a new role for Aβ: fighting viruses. The reason our brains have Aβ in the first place is somewhat unclear, and according to this study from Dr. Robert Moir’s group at Harvard, Aβ forms clusters in response to viral infection and the clusters help to trap the virus particles so that they cannot attach to brain cells [5]. The theory further suggests that if a latent virus becomes active again due to age and environmental stressors, the brain might respond by increasing production of Aβ clusters, starting the amyloid cascade.
This is particularly interesting because it suggests that the clustering property of Aβ might have a functional purpose rather than simply being a pathological symptom of AD. I like the idea. It brings a little depth of character to the demonized Aβ. However, while the study is intriguing, take it with a grain of salt. You don’t need to be terrified if you’ve recently had a viral infection. Certainly not all cases of AD arise from an infection, and the authors explicitly admit, “Our findings do not constitute direct evidence of a role for infection in AD etiology.” Rather, the results point to infection as new and perhaps underappreciated risk factor, one that merits further research.
Skin for the Cure
So does all this new research mean we’re on the brink of a cure? Not quite. While the recent findings enumerated above have made splashes in the field, the past few months have not been particularly unusual in the AD world. Research is generated so quickly that it is virtually impossible to read all the results, and yet time drags on without a cure. Why? The problem is two-fold (well probably multi-fold but there are two really important folds). First, clinical trials for any new drug are extremely risky given the exorbitant cost, so very few findings in the lab reach clinical tests. Second, while mouse models have helped us learn so much about AD, they are not perfect and generate a lot of false positives. This bottleneck inherent in the progression from basic research to clinical relevance is intensely frustrating.
A possible solution? Skin! Our stem cell technology is reaching science fiction levels. Scientists are now able to take a skin biopsy from an AD patient, turn the skin cells into stem cells, and then make the stem cells turn into neurons. These neurons in a dish remarkably develop marks of AD pathology. This technique provides an incredible potential for drug screening and even personalized medicine. Testing drugs using these skin-derived neurons is quick and cheap and allows scientists to vastly increase the number of drugs that they are able to test. If this drug-screening method lives up to its potential and if researchers continue to pursue novel ideas and strategies for fighting AD, a cure might be on the horizon.
References
- Katzman R, Terry R, DeTeresa R, et al. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Annals of neurology. 1988 Feb;23(2):138–44
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016 May 6;352(6286):712-6. doi: 10.1126/science.aad8373. Epub 2016 Mar 31. PubMed PMID: 27033548.
- Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL, Duff KE. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med. 2016 Jan;22(1):46-53. doi: 10.1038/nm.4011. Epub 2015 Dec 21. PubMed PMID: 26692334; PubMed Central PMCID: PMC4787271.
- Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA, Herman M, Nahmani E, Emrani S, Figueroa YH, Diamond MI, Clelland CL, Wray S, Duff KE. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016 Aug;19(8):1085-92. Doi: 10.1038/nn.4328. Epub 2016 Jun 20. PubMed PMID: 27322420; PubMed Central PMCID: PMC4961585.
- Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016 May 25;8(340):340ra72. doi: 10.1126/scitranslmed.aaf1059. PubMed PMID: 27225182.

You must be logged in to post a comment.