 November
15
November
15
Tags
From symptoms to biology: shifting definitions of Alzheimer’s disease
As a neuroscientist studying Alzheimer’s, I’m reminded of its far-reaching impact each time a barista, cashier, or Lyft driver makes small talk by asking what I do for a living. Unfortunately, this devastating disease needs no introduction. Considering its ubiquity, it’s surprising that a debate broke out recently among leaders in the field over the definition of Alzheimer’s disease (AD). It’s not as if scientists are just getting around to defining AD, so why is this debate happening now?
Then and Now
Let’s back up all the way to when the disease got its name. The first patient officially diagnosed with Alzheimer’s disease was a woman with dementia named Auguste Deter. Upon her death in 1906, Dr. Alois Alzheimer examined her brain and described the presence of abnormal clumps of protein outside of neurons (amyloid plaques) and twisted strands of protein inside of neurons (tau tangles). These protein abnormalities–the plaques and tangles–became the hallmark pathological changes of AD. Unable to examine their living patients’ brain tissue, doctors diagnosed probable Alzheimer’s dementia in people with specific symptoms, such as memory loss. This clinical diagnosis could be confirmed (becoming a definitive diagnosis) upon the discovery of plaques and tangles after death.
However, recent advances in neuroscience have led to the development of tools that allow researchers to assess the amount of these abnormal proteins in living people. We can also assess the extent of neurodegeneration (the progressive loss of neurons), which, while not specific to AD, is very closely related to the cognitive decline seen in AD and other forms of dementia. In light of these advances, a recent set of guidelines [1] suggests that researchers should shift their definition of AD diagnosis away from its clinical symptoms and back toward the original, biological definition of the disease–the presence of plaques and tangles–allowing for a definitive diagnosis to be made while a patient is alive.
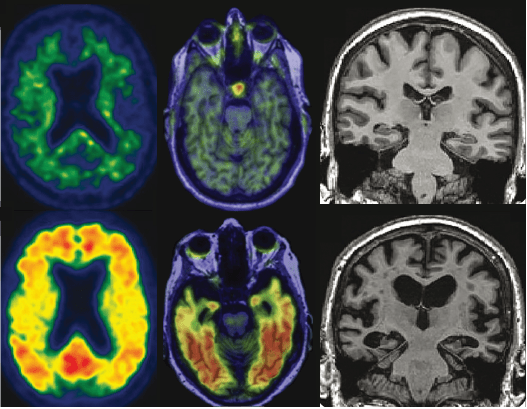
Examples of normal (top row) and abnormal (bottom row) amyloid, tau, and neurodegeneration as seen with PET and structural MRI
Easy as AT(N)
In the new framework, information about levels of amyloid (A) and tau (T) are used to stage patients along the AD continuum, while neurodegeneration (N) is used to stage disease severity. Each biomarker (the indicator of amyloid, tau, or neurodegeneration) is binarized into either positive or negative, leading to eight AT(N) biomarker profiles. Essentially, the presence of multiple abnormal (positive) biomarkers is indicative of more advanced disease.

Because of a prevailing theory which suggests that amyloid buildup is the crucial, early event in AD, any profile that includes A- is considered either normal or possessing another non-AD pathology. If A+, a person is now firmly on the AD continuum and will fall into a different biomarker category depending on the status of the other two biomarkers (T&N). For example, if a person is A+T-N-, the category is AD pathologic change (the most mild), whereas A+T+ is considered Alzheimer’s disease, regardless of N status. Current evidence suggests the order in which these biomarkers would become abnormal in AD is A -> T -> N -> cognitive impairment, but other pathways are still being tested.
What isn’t included in AT(N)? Clinical symptoms. Each of these biomarker categories is independent of cognitive status–whether you’re cognitively normal or have dementia, it won’t be reflected in your AT(N) profile. If an otherwise seemingly healthy individual is A+T+ they’re considered to have preclinical Alzheimer’s disease, meaning the abnormal protein hallmarks are present and precede the cognitive changes, like memory impairment, we associate with AD. The idea of preclinical AD isn’t new–in fact, the concept began with the observation that some non-demented older adults had AD pathology at autopsy [2]. Research has since shown that Alzheimer’s pathology begins to accumulate decades before clinical symptoms appear.
Researchers in favor of the new framework consider the symptoms to be a consequence of the disease and say it’s critical to focus on the underlying biological changes. After all, potential treatments that target the hallmark plaques and tangles should only be given to patients who actually have those abnormalities. It sounds obvious, but this type of work was inconceivable before we were able to visualize AD pathology in the brains of living patients because some individuals diagnosed with AD based on symptoms alone actually have a different kind of pathology in their brains. Now, new lines of research–such as the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s study–seek to test whether decreasing amyloid in cognitively healthy, amyloid-positive individuals will slow the development of AD-related memory loss.
So, what’s the issue?
Biomarkers are so frequently used in research and clinical settings that it certainly seems logical that AD would follow suit with AT(N). After all, it’s commonplace for a doctor to test your LDL (“bad”) cholesterol to assess your risk of heart disease. If it’s high, lifestyle changes and drugs like statins can lower your LDL, which in turn helps prevent heart disease. The premise is simple. But to understand some of the issues surrounding a biomarker-based definition of AD, we first have to understand what biomarkers are and how they’re evaluated more generally.
What makes a valid biomarker? A biomarker is a measurable, objective indication of the presence or severity of a disease. It should be sensitive enough to identify those with a disease and specific enough to identify those without it. It should also accurately predict clinical outcomes across diverse populations and be reproducible across research labs or clinics.
In AD, researchers have yet to agree on how to standardize the methods of assessing the extent of pathology or how to determine if an individual is positive or negative on a given test result. Let’s use amyloid as an example. Amyloid can be measured with PET imaging as shown above, but it can also be measured in cerebrospinal fluid (CSF), the water-like liquid that bathes the central nervous system, which can be sampled with a spinal tap. Within either method, there are additional choices (there’s no standard way to measure amyloid in the CSF or kind of PET to use (PiB? Florbetapir? Florbetaben?)), including where and how to set the cutoff for being considered positive or negative for amyloid. Such tests haven’t even been validated outside of the largely white, wealthy, and well-educated subjects who make up most research cohorts, meaning the cutoffs we currently have may not apply to the majority of people anyway. To complicate things more, these two methods don’t actually measure the same thing. CSF is more like a snapshot in time of the current state, whereas PET shows the accumulated amyloid up until this point. And then there’s the issue that both CSF and PET measures estimate a certain form of amyloid (the fibrils, or aggregates of stuck together proteins which accumulate in plaques) when it’s thought that a different, soluble form of amyloid may actually be the toxic kind.
In moving toward a biomarker-based definition, leaving cognition out of the equation means that seemingly healthy older adults–without any memory problems–would now find themselves on the AD continuum. AD has come to be synonymous with a syndrome of memory impairment. What would it mean to have AD (according to the biological definition) without any symptoms? Most of the concern when it comes to AD is about the cognitive change: the memory loss, the debilitating disruption of daily life. A complicated twist to our newfound ability to identify AD pathology in living people is the discovery that most people with preclinical Alzheimer’s will never develop dementia in their lifetime [3]. It may seem counterintuitive–after all you might’ve heard that an estimated 1 in 3 seniors dies with AD or another dementia, or that AD is the 5th leading cause of death for those 65 and older. In fact, research indicates that up to 65%(!) of individuals in their 80s or older are amyloid positive [4]. However, in this slowly developing disease, an individual may not live long enough to experience the cognitive changes associated with these brain abnormalities.
Another important wrinkle is that, so far, there’s no evidence that reducing AD pathology improves cognition. If you recall the heart disease example, a key component to the use of LDL cholesterol as a biomarker is that by lowering LDL, the risk for heart disease is reduced. Our current theory of amyloid in AD suggests the same: by lowering amyloid, the risk for AD is reduced. The best evidence in support of our current model of AD would come from successfully removing amyloid from the brain and consequently witnessing a measurable change in cognitive decline [5], but that hasn’t happened yet. In fact, clinical trials attempting to do just that have failed to slow disease progression despite reducing amyloid levels. It’s possible that treatments haven’t worked because they need to be given earlier in the course of disease, but it’s also clear we don’t fully understand the biological relationship between amyloid, tau, neurodegeneration, and cognitive decline in AD, and are just starting to realize the importance of other factors, like vascular disease and inflammation.
Looking ahead
Proponents of the AT(N) framework acknowledge there is a lot of work to be done and emphasize that, at least for now, this definition only applies to research, not clinical practice. But critics feel this shift is at best premature and worry that once such a framework is established for research, it’ll be carried over into the clinic. Given the media coverage of the new framework, it’s reasonable to assume that concerned patients will want to get that Alzheimer’s scan they read about in the paper, and the ethics of disclosing biomarker status to patients is still being explored. Still, in the current era of big data where research collaborations stretch across institutions around the globe, an objective, measurable definition would ensure we’re communicating about the same thing when we say “Alzheimer’s” so that results can readily be compared between groups.
The new framework is certainly imperfect, and will need to be refined with scientific advances. Still, many are hopeful that efforts to target AD as a distinct biological entity will lead to the development of a targeted therapy that slows, halts, or even prevents this disease from progressing.
References
- Jack, C., Bennett, D., Blennow, K., Carrillo, M., Dunn, B., Haeberlein, S., Holtzman, D., et al. (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & Dementia, 14(4), 535–562.
- MANN, TUCKER, & YATES. (1987). THE TOPOGRAPHIC DISTRIBUTION OF SENILE PLAQUES AND NEUROFIBRILLARY TANGLES IN THE BRAINS OF NON‐DEMENTED PERSONS OF DIFFERENT AGES. Neuropathology and Applied Neurobiology, 13(2), 123–139.
- Brookmeyer, R., & Abdalla, N. (2018). Estimation of lifetime risks of Alzheimer’s disease dementia using biomarkers for preclinical disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association.
- Rowe, C., Ellis, K., Rimajova, M., Bourgeat, P., Pike, K., Jones, G., Fripp, J., et al. (2010). Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiology of Aging, 31(8), 1275–1283.
- Jagust, W. (2018). Imaging the evolution and pathophysiology of Alzheimer disease. Nature reviews Neuroscience, 19(11), 687–700.
Title image by Anita Jankovic
Additional images adapted from [1] and Jack, C. R., Bennett, D. A., Blennow, K., Carrillo, M. C., Feldman, H. H., Frisoni, G. B., Hampel, H., Jagust, W. J., Johnson, K. A., Knopman, D. S., Petersen, R. C., Scheltens, P., Sperling, R. A., … Dubois, B. (2016). A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology, 87(5), 539-47.

You must be logged in to post a comment.