 April
21
April
21
Tags
The Genetic Curse of Fatal Insomnia
Not being able to fall asleep when you want to is frustrating. No matter how tired we are, we all have nights when we just can’t fall or stay asleep. The role of sleep in our everyday health is critical, which is no more apparent than the day after one of those sleepless nights when we might feel grumpy, forgetful, or ill.
What if you were, however, gradually unable to fall asleep? In very rare cases of people with fatal familial insomnia (FFI), this is exactly the outcome of their genetically-driven neurodegenerative disease.
Fatal Familial Insomnia
FFI is a genetically-acquired disease which is, as the name suggests, marked by the inability to fall asleep. Sadly, the disease is always fatal and follows an accelerated course of degeneration – by the time patients begin to show insomnia symptoms and are diagnosed in their 40s, 50s, or 60s, FFI only takes an average of 18 months to cause death [1,2,3]. To assuage the fears of any insomniacs reading this article, cases of FFI are extremely rare, with only 70 total families worldwide ever reported in the scientific literature to pass down the genetic defect [4]. FFI is caused by a mutation in a gene called PRNP, which encodes a protein called prion protein (more on this later). It is inherited through family genetics in an autosomal dominant manner, meaning just one mutated copy of the two PRNP genes – one inherited from each parent – leads to disease. Therefore, children have a 50% chance of inheriting the disorder when one of their parents carries the mutation [4,5].
In addition to mild insomnia that subsequently develops into the total inability to fall asleep, patients typically experience other mental and physical symptoms as the disease progresses. Psychological effects include confusion, dementia, hallucinations, and issues with memory and language. Physically, FFI patients may experience trouble with the coordination of movement, tremors, muscle spasms, and difficulty moving their eyes and swallowing. Other hallmarks of the disease follow from dysregulation of the autonomic nervous system, which is made up of the nerves that control involuntary bodily functions. This dysregulation leads to rapid heart rate, sweating, pupil dilation, and inability to regulate body temperature [5].
So, how does a genetic defect and its effect on the brain cause these devastating effects? What can we learn about the reciprocal relationship between sleep and the brain? And is there any hope for treatment?
What causes brain degeneration in FFI?
FFI is known as a prion disease. Prion diseases are caused by a mutation in the genetic code for a specific protein called the prion protein, which causes it to misfold. The ability of proteins to do their jobs in our body relies heavily on their structure – after a genetic sequence is translated into protein building blocks, these building blocks must fold into the correct shape. Certain genetic mutations, such as the one that causes FFI, disrupt the ability of that protein to take on the correct shape. Although it’s not understood how this happens, the misfolded prion protein can induce other proteins to take on an inappropriate shape. This not only makes the proteins unable to proceed with their usual roles, but can also cause them to stick to each other in clumps [6].

You may have heard of prion diseases in the news if you’ve ever paid attention to outbreaks of “mad cow disease.” Yes, prion diseases are actually transmissible. Ingestion or injection of the misfolded protein can cause the misfolding of proteins in another host. While “mad cow disease” circulated among, well, cows, outbreaks of another disease – variant Creutzfeldt-Jakob Disease – appeared in humans and is thought to have occurred from humans ingesting the meat from prion-producing cows [7, 8]. Similarly to FFI, there has sadly never been a human survivor of this type of prion disease. Kuru, another prion disease found in certain tribes in New Guinea, continued its transmission through human populations by the ritual funeral ingestion of deceased prion carriers’ brains [9].
In the case of FFI, which is a genetic condition, patients don’t acquire the misfolded protein from another source. They actually make the prion themselves due to the faulty genetic code. Most often, this occurs through familial genetic inheritance, but extremely rarely it can occur in other individuals through random genetic mutation. In theory, FFI might also be transmissible. This transmission would probably require ingestion of prion-affected brain tissue itself, but other prion diseases have also been theorized to transmit very rarely during medical procedures like blood transfusion or organ transplants as the prions can be found in other bodily tissues at very low levels. Interestingly, scientists aren’t exactly sure what the normal, regularly folded role of human prion protein is in healthy individuals, which is why the gene and protein are named after the problem prions. Some roles of the healthy protein that have been proposed include its playing a part in signaling between cells and protecting brain cells from injury [6]. Perhaps even more importantly for FFI, a study in which the healthy version of the prion protein was genetically deleted in mice indicated that a lack of the healthy protein (even in the absence of the mutated, disease causing protein) affected sleep patterns and quality [10].
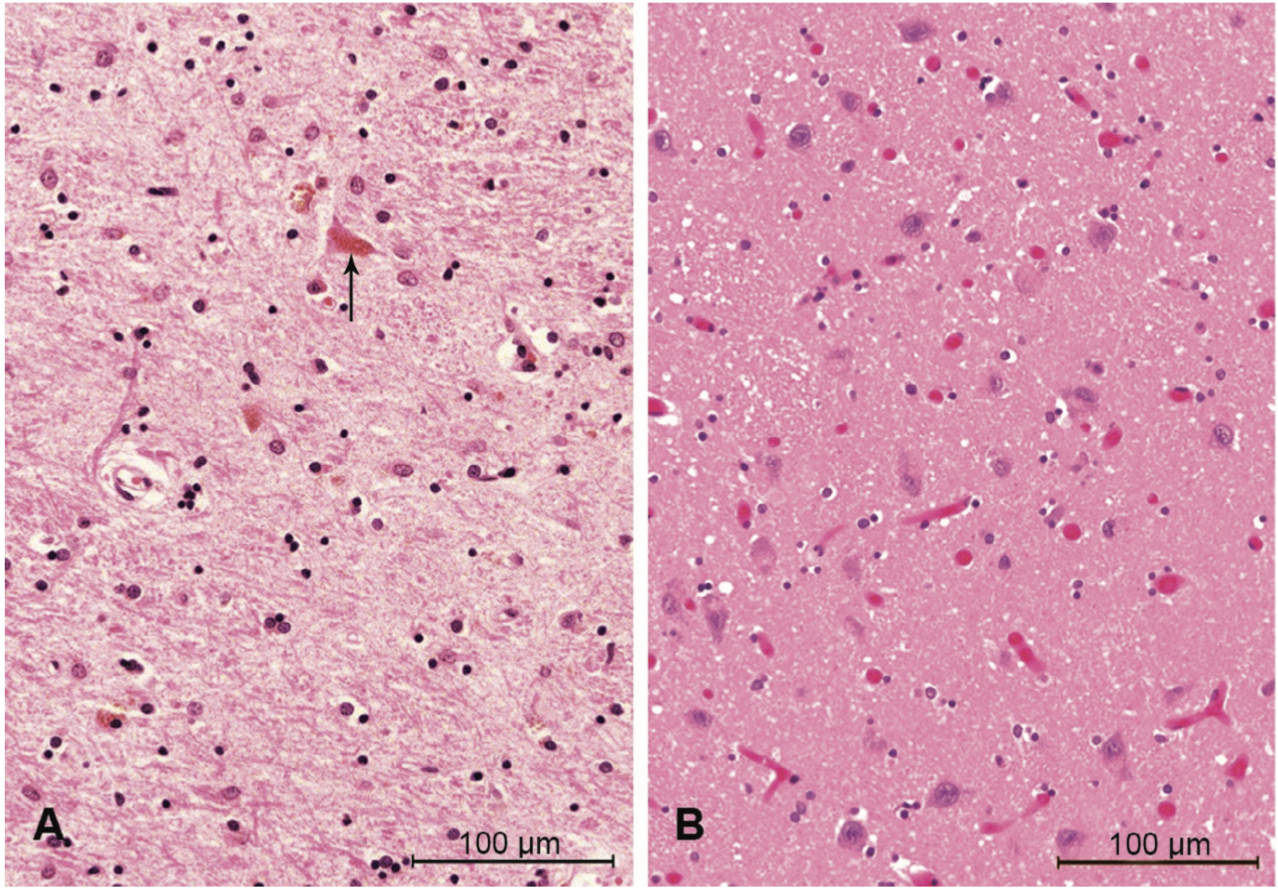
This idea of clumped proteins may make you think of another common neurodegenerative disease – Alzheimer’s disease – where at least part of the dementia is thought to be caused by inappropriate clumps of proteins that are toxic to neurons and lead to their destruction, especially in the parts of the brain important for memory. Alzheimer’s disease is not categorized as a prion disease, however, because the effects are not mediated by the same prion protein. Nonetheless, FFI works similarly – clumps of proteins are found particularly in an area of the brain called the thalamus, which has many functions that will be discussed in subsequent sections [1]. The clumps of prion protein lead to the total destruction of neurons in this brain area, which contributes to most of the symptoms described above.

FFI, the thalamus, and sleep
The major hallmark of FFI is the total degeneration of the thalamus. Located in the center of the human brain, the thalamus is the major hub that routes incoming sensory information to destinations all over the brain, especially to the cerebral cortex. Due to its super interconnected nature with other parts of the brain, a healthy thalamus is implicated in many different types of perception and behavior. Destruction of the thalamus can therefore lead to movement and thought dysfunction, as well as autonomic dysfunction, probably through its connections with the nearby hypothalamus. Importantly for understanding FFI, the thalamus has also been implicated in sleep and wakefulness cycles by coordinating the large-scale waves of electrical activity that travel across the brain and are indicative of sleep [11,12]. Interestingly, stroke damage to the thalamus can cause permanent coma, while prion damage in the thalamus in FFI leads to persistent wakefulness. But what is so important about sleep that its absence would lead to death?
Thus far, scientists have been able to identify a variety of vital roles sleep plays in maintaining our physical and mental well-being. It is thought that during sleep, toxins that have built up in the brain throughout the day are removed and memories are consolidated for future use [13]. Sleep cycles through a variety of stages each night that each exhibit specific brain activity (for a detailed look at sleep stages, check out these previous NeuWriteSD articles). Deep, restorative, slow wave sleep transitions to REM (rapid eye movement) sleep, which is when we mostly experience dreaming. Patients with FFI first lose the ability to enter slow wave sleep, and eventually experience the loss of REM sleep. When electroencephalography (EEG) is used to examine electrical activity in the brains of healthy and FFI patients via electrodes placed on the scalp during sleep, FFI patients do not show any of the expected brain waves controlled by the thalamus that signify sleep. In fact, their brain activity is not consistent with wakefulness either, leaving patients in a persistent state somewhere between wake and sleep. Further into disease progression, any lapse into a REM-like state that does occur becomes unlinked from sleep, leaving patients in a stupor and acting out waking dreams at any point in the day [14].
You may be asking yourself, what is the fatal component of FFI? Does the prion-induced cell death and brain damage lead to death? Or is it the inability to sleep? Some researchers and doctors treating FFI patients think it may be a combination of the two. Certainly, prion-induced damage will eventually spread to enough of the brain such that important bodily functions will cease. However, one group of researchers suggest that post-mortem analysis of brain tissue indicates that FFI brains don’t appear to have enough damage to make this the sole cause of death [3]. Sleep deprivation in human subjects in laboratory studies has not been shown to be fatal itself (of course, the researchers would not allow a study to progress to the point of death, but one 1960s case study did follow a patient who remained awake for 264 hours with no lasting effects after recovery sleep [15]), but it is fatal in some species of animals. Likely, patients perish due to the combination of brain damage, lack of sleep’s clearance of brain and body toxins, and the inability to access sleep to gain relief from autonomic hyperdrive (the increased heart rate, sweating, pupil dilation, etc.) that can cause the body to burn out.

Is there hope for a cure?
Fatal familial insomnia is an extremely devastating genetic and neurodegenerative disease for which there is no current cure. While some treatments, such as certain antibiotics that are thought to prevent the prion protein from clumping, are currently undergoing trials in animals and humans, the preliminary results haven’t been strong [16]. Physicians often opt for medical interventions that can help alleviate individual symptoms of the disease, although a majority of FFI patients do not respond to sedatives that are meant to induce rest and sleep. In one case study, an intense regimen of vitamins, sedatives, sensory deprivation and other therapies designed to promote sleep did enhance the length and quality of life of one patient by one year [17]. While this does indicate that lack of sleep itself is a crucial player in disease progression, these therapies are still unable to mitigate the prion-driven brain degeneration that causes death. A major obstacle to clinical trials in human patients is that medical interventions likely need to start before there is enough thalamic damage to cause symptoms. In this case, patients would need to undergo genetic testing for the disease-causing mutation. Understandably, many individuals from FFI families would simply rather not know if their future will include an invariably fatal disease. Would you?
References
- Llorens F, Zarranz JJ, Fischer A, Zerr I, Ferrer I (2017) Fatal familial insomnia: clinical aspects and molecular alterations. Current Neurology and Neuroscience Reports, 17:30
- Cracco L, Appleby BS, Gambetti P (2018) Fatal familial insomnia and sporadic fatal insomnia. Handbook of Clinical Neurology, 153:271-299
- Schenkein J, Montagna P (2006a) Self management of fatal familial insomnia. Part 1: what is FFI? Medscape General Medicine, 8(3): 65
- National Institutes of Health Genetic and Rare Diseases Information Center. Fatal Familial Insomnia, https://rarediseases.info.nih.gov/diseases/6429/fatal-familial-insomnia
- National Organization for Rare Disorders (2018) Fatal Familial Insomnia. https://rarediseases.org/rare-diseases/fatal-familial-insomnia/
- MedlinePlus [Internet]. Bethesda (MD): National Library of Medicine (US). Prion disease; [updated 2020 Aug 18; reviewed 2014 Jan 1; cited 2022 Apr 21]. Available from: https://medlineplus.gov/genetics/condition/prion-disease/
- Centers for Disease Control and Prevention, US Department of Health and Human Services [Internet]. Atlanta (GA). Variant Creutzfeldt-Jakob Disease; [updated and reviewed 2022 Jan 28; cited 2022 Apr 21]. Available from: https://www.cdc.gov/prions/vcjd/index.html
- United States Food and Drug Administration [Internet]. Silver Spring (MD). All about BSE (mad cow disease); [updated and reviewed 2020 Jul 23; cited 2022 Apr 21]. Available from: https://www.fda.gov/animal-veterinary/animal-health-literacy/all-about-bse-mad-cow-disease
- MedlinePlus [Internet]. Bethesda (MD): National Library of Medicine (US). Kuru; [updated 2022 Apr 1; cited 2022 Apr 21]. Available from: https://medlineplus.gov/ency/article/001379.htm
- Tobler I, Gaus SE, Deboer T, Achermann P, Fischer M, Rulicke T, Moser M, Oesch B, McBride PA, Manson JC (1996) Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature, 380: 639-642
- Gent TC, Bassetti CLA, Adamantidis AR (2018) Sleep-wake control and the thalamus. Current Opinion in Neurobiology, 52:188-197
- Gent TC, Bandarabadi M, Gutierrez Herrera C, Adamantidis AR (2018) Thalamic dual control of sleep and wakefulness. Nature Neuroscience, 21:974-984
- Sara SJ (2017) Sleep to remember. Journal of Neuroscience, 37(3):457-463
- Montagna P, Cortelli P, Avoni P, Tinuper P, Plazzi G, Gallassi R, Portaluppi F, Julien J, Vital C, Delisle MB, Gambetti P, Lugaresi E (1998) Clinical features of fatal familial insomnia: phenotypic variability in relation to a polymorphism at codon 129 of the prion protein gene. Brain Pathology, 8:515-520
- Ross, JJ (1965) Neurological findings after prolonged sleep deprivation. Archives of Neurology, 12:399-403
- Lavigna G, Masone A, Bouybayoune I, Bertani I, Lucchetti J, Gobbi M, Porcu L, Zordan S, Rigamonti M, Imeri L, Restelli E, Chiesa R (2021) Doxycycline rescues recognition memory and circadian motor rhythmicity but does not prevent terminal disease in fatal familial insomnia mice. Neurobiology of Disease, 158:105455
- Schenkein J, Montagna P (2006b) Self management of fatal familial insomnia. Part 2: case report. Medscape General Medicine, 8(3): 66
Image Sources
Prion Protein Structure: AtikaAtikawa/Wikimedia Commons/CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=86424297
Tractography: Yeh, F. C., Panesar, S., Fernandes, D., Meola, A., Yoshino, M., Fernandez-Miranda, J. C., … & Verstynen, T. (2018). Population-averaged atlas of the macroscale human structural connectome and its network topology. NeuroImage, 178, 57-68. – http://brain.labsolver.org/, Wikimedia Commons/ CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=78298711
Cover Image: Wodgester/Wikimedia Commons/CC BY-SA-4.0

You must be logged in to post a comment.