 July
05
July
05
Tags
Where does Alzheimer’s disease begin?
Sometimes I forget what day of the week it is, where I put my keys, or when a friend’s birthday is- but I never stop to wonder if these brief moments of forgetfulness are normal or a sign of something more serious. For many, occasional short-term memory loss is a normal part of getting older, and often isn’t given much concern. However, this also happens to be one of the earliest symptoms of Alzheimer’s disease (AD). Over time, the symptoms get progressively worse. People with AD often experience difficulty in carrying out their daily tasks, changes in their mood and personality, and even trouble with speaking and walking.
Unfortunately, by the time the symptoms are severe enough to suspect a diagnosis of AD, irreversible loss of cells and their connections has already occurred. This is one of the biggest challenges facing researchers trying to treat this disease; insidious changes start taking place in the brain decades before people with AD start showing severe impairments. If the earliest signs of AD are often mistaken as signs of healthy aging, how can we better detect these signs earlier in life to prevent widespread brain damage from happening?
Recently, evidence has suggested that AD pathology might not begin where we thought it did (near the hippocampus, a part of the brain important for learning and memory). Rather, subtle changes might start taking place much earlier, perhaps decades, in the brainstem (which is broadly involved with alertness, awareness, and the sleep cycle, among other things). If this is true, there may be a therapeutic window of opportunity to treat AD patients before irreversible damage occurs.
Some Background
First, let’s briefly discuss what we already know about AD pathology. AD is characterized by the presence of abnormal clusters of two molecules: amyloid beta (Aβ) and tau. These molecules are found in the healthy brain as well, but in AD, they aggregate into clusters that aren’t cleared out as they should be. Clusters of Aβ form outside of neurons, and are called plaques. Clusters of tau form within neurons, and are called tangles. As these plaques and tangles accumulate, the connections between neurons as well as the neurons themselves are damaged and start to die. This results in atrophy (or shrinkage) of the brain, leading to many of the devastating symptoms we see in patients with AD (see Catie’s post for more background information on AD!).

“Alzheimer disease is characterized by brain atrophy associated with loss of synapses and neurons. At the microscopic level, deposition of extracellular amyloid-β plaques and intraneuronal neurofibrillary tangles is observed, in association with dystrophic neurites and loss of synapses, as well as microgliosis and astrogliosis.” | source: https://www.nature.com/articles/s41582-018-0013-z.pdf
So far, clinical trials for treating AD have mostly focused on removing or preventing the formation of Aβ plaques. However, these attempts have been largely unsuccessful, so more attention is shifting toward tau-based therapies.
Tau pathology in AD
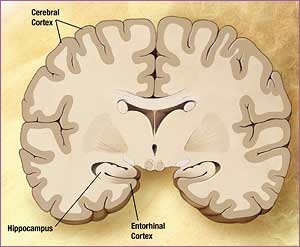
source: http://editthis.info/psy3242/Entorhinal_cortex
So what do we know about the progression of tau pathology in AD? In 1991, Heiko and Eva Braak (1) proposed a system to divide the progression of AD into stages of severity based on tau pathology. They examined postmortem brains from people with and without AD, and found abnormal tau first in the entorhinal cortex, a region that connects the hippocampus with other parts of the brain (stages I-II). Next, they saw abnormal tau in limbic areas, including the hippocampus (stages III-IV). Finally, brains in the most advanced stages of AD showed widespread irregular tau throughout the cerebral cortex (stages V-VI). This pattern parallels the timeline of symptoms we see- beginning with subtle memory impairments, followed by more severe memory impairments with changes in mood and behavior, and finally, changes in personality, decision-making, and other higher cognitive functions.
However, in 2011, Heiko Braak (2) revised his original theory. This time, he examined a larger number of brains from a wider age range of people, and concluded that tau pathology may actually begin in the lower brainstem, in a region called the locus coeruleus (LC). He revised his original staging system, coining these first LC changes as stage 0. He suggested that the pathologic tau changes then spread from the LC into the entorhinal cortex, where it follows the classic staging pattern.

LC in blue | source: shutterstock.com
The LC is the brain’s main source of norepinephrine, a chemical involved in attention and arousal, the sleep cycle, and memory, among other things. Needless to say, degeneration of this area has widespread effects on behavior and cognition. Since Braak revised his original hypothesis, scientists have found that higher density of neurons in the LC protects against cognitive decline in aging, and a greater number of tau tangles in the LC correlates with greater cognitive decline (3,4). Further, this hypothesis could explain why people with AD often show early impairments in non-cognitive areas like sleep and mood.
What implications does this have for the study of AD? As mentioned earlier, one of the biggest struggles we face in treating the disease is that we aren’t able to catch it early enough to prevent the destruction of cells. If tau pathology begins in the brainstem long before we see damage in the entorhinal cortex, we might be able to target therapies toward the locus coeruleus cells earlier in life to prevent neurodegeneration.
New avenues for treatment
In 2017, Rorabaugh and colleagues (5) introduced a rat model of AD (called TgF344-AD) which has disease-causing mutations in genes related to the production of amyloid (the molecule which makes up “plaques”). Interestingly, this rat model showed accumulation of abnormal tau in the LC much earlier than in the entorhinal and cortical areas, and before any actual degeneration. This demonstrated that these rats are a good model to study the effects of early LC pathology on the progression of AD.
To do just that, the authors tested these rats along with healthy controls in the Morris water maze task. This is a commonly used test in neuroscience research to study learning and memory. In this task, animals are placed in a tank filled with opaque water. Somewhere in the tank, a platform is hidden a couple of centimeters below the water. The rats are given many trials to learn the location of the platform. In this experiment, the AD rats took longer to find the platform than the healthy rats, suggesting that they had deficits in spatial learning and memory. This supports the idea that this particular type of rat is a good model for AD.
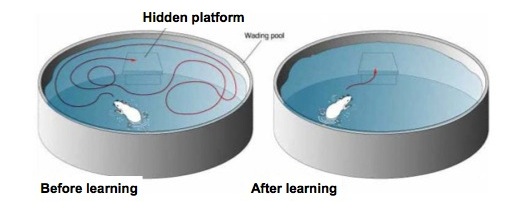
source: http://mikeclaffey.com/psyc170/notes/notes-memory.html
Perhaps the most exciting finding of this study was that the authors were able to restore the ability of the AD rats to learn the platform location just as well as the healthy controls. They used a chemogenetic technique called designer receptors exclusively activated by designer drugs (DREADDs) to activate neurons in the LC, and afterwards, the AD model rats no longer took more time than the healthy rats to learn the location of the platform. These findings suggest that there is a window of opportunity to treat AD by using norepinephrine-based therapeutics targeted at the LC, but this treatment has to happen before irreversible cell loss.
The future of AD research
So, where does this leave us? We have evidence that AD may actually begin in the LC of the brainstem rather than in the entorhinal cortex, and the changes that take place begin much earlier (perhaps decades in humans) before clear cognitive impairments. Rorabaugh et al. (2017) found that increasing LC activity before degeneration improved AD rats’ ability to complete a spatial learning task, suggesting that we might be able to use therapeutics targeted at the LC to alleviate AD symptoms, or even prevent the disease from progressing.
Exciting as this is, we still don’t know how to tell which people will go on to develop AD early in life, nor do we know when the optimal time to administer an LC-targeted treatment in humans may be. Still, this is a compelling and novel avenue of research for AD scientists, and has a lot of promise for the treatment of the disease.
REFERENCES
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82(4):239-59.
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories From 1 to 100 Years. J Neuropathol Exp Neurol 2011; 70(11): 960-969.
- Wilson RS, Nag S, Boyle PA, Hizel LP, Yu L, Buchman AS, et al. Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology 2013; 80: 1202–8.
- Kelly SC, He B, Perez SE, Ginsberg SD, Mufson EJ, Counts SE. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol Commun 2017; 5: 8.
- Rorabaugh JM, Chalermpalanupap T, Botz-Zapp CA, Fu VM, Lembeck NA, Cohen RM, et al. Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer’s disease. Brain 2017; 140: 3023–38.

You must be logged in to post a comment.